№ 2, 2002 г. № 2, 2002 г.
№ 2, 2002 г. № 2, 2002 г.© В.М.РойхельМедленные болезни
человека и животных,
вызванные прионамиВ.М.Ройхель
Виктор Моисеевич Ройхель, д. м. н., ведущий научный сотрудник
Института полиомиелита и вирусных энцефалитов им.М.П.Чумакова РАМН.Эта группа заболеваний прославилась после разразившейся в Великобритании эпизоотии губкообразной энцефалопатии крупного рогатого скота, больше известной под названием «коровье бешенство». К 1997 г. было инфицировано около 1 млн голов крупного рогатого скота, примерно 54 тыс. инфицированных животных попали в пищевую цепь людей [1]. Особенно был напуган мир, когда появились сведения о возможной связи между этим заболеванием и возникновением нового варианта болезни Крейтцфельдта-Якоба - смертельной инфекции человека, которая, в отличие от ее классического варианта, поражает молодых людей, как правило, в возрасте 27 лет [2, 3].
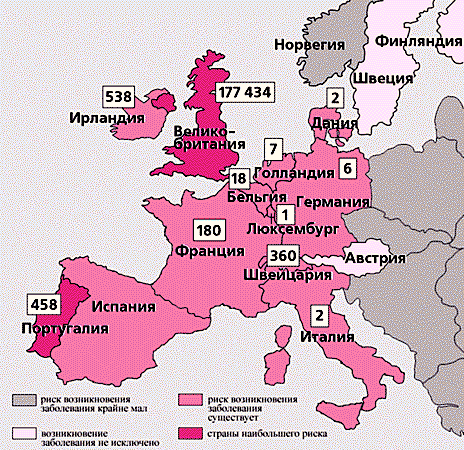
Случаи (указаны числами) «коровьего бешенства» среди сельскохозяйственных животных и риск заражения человека болезнью Крейтцфельдта-Якоба в некоторых европейских странах. Риск появления этого заболевания в США и Канаде маловероятен, но не исключен. В остальных странах опасность инфекции крайне мала.Нет ничего удивительного, что эти события вызвали повышенный интерес специалистов к новому типу инфекции человека и животных. Активизировались поиски, а затем изучение весьма своеобразного возбудителя, принципиально отличающегося от всех известных инфекционных агентов - вироидов, вирусов, бактерий и простейших. Хотя в России нет реальной угрозы массового заражения возбудителем этого экзотического заболевания, тем не менее биомедицинские исследования ведутся и у нас.«История болезни»
Понятие «медленные болезни» впервые ввел исландский исследователь Б.Сигурдссон еще в 1954 г., который и сформулировал основные характерные черты этой особой формы инфекции: поражение одного органа или одной системы и наличие одного хозяина, продолжительный инкубационный период (от нескольких месяцев до нескольких лет), неуклонное нарастание клинической симптоматики, неизбежно приводящее к смерти [4]. Сигурдссон изучал медленные болезни только животных (в частности, подробно исследовал распространенное во всем мире заболевание овец - скрепи), однако вскоре выяснилось, что они могут поражать и человека. В 1957 г. американский ученый К.Гайдушек описал новое заболевание - куру, выявленное у папуасов-каннибалов, жителей Новой Гвинеи. Болезнь носила массовый характер, и вскоре была доказана ее инфекционная природа. За эти исследования Гайдушек был удостоен в 1976 г. Нобелевской премии.
Возбудителями медленных болезней, как впоследствии выяснилось, могут стать хорошо известные «виновники» острых инфекций: например, вирусы кори, краснухи, герпеса, гриппа, клещевого энцефалита и др. (всего около 40 болезней). Что произойдет в организме - медленное и фатальное течение инфекционного процесса или острое - зависит от условий, в которых оказался возбудитель [5].
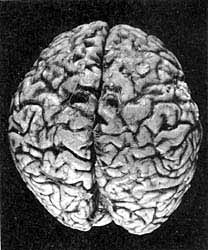
Головной мозг человека, погибшего от болезни Крейтцфельдта-Якоба. Видны явные патоморфологические изменения: уменьшение объема и массы мозга, истончение извилин полушарий большого мозга, преимущественно лобных и теменных долей со значительным расширением борозд в этих областях. (В.А.Зуев, И.А.Завалишин, В.М.Ройхель, 1999.)
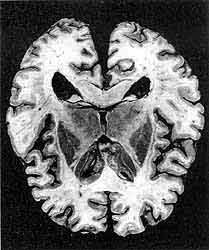
Горизонтальный срез головного мозга человека, умершего от спорадической формы болезни Крейтцфельдта-Якоба. Заболевание привело к сужению коры мозга в лобной, теменной, височной и затылочной долях, а также произошло некоторое уменьшение объема базальных ядер и таламуса и умеренное расширение желудочков мозга. (В.А.Зуев, И.А.Завалишин, В.М.Ройхель, 1999.)
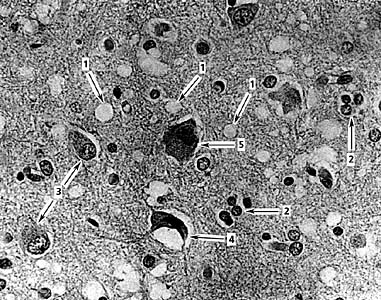
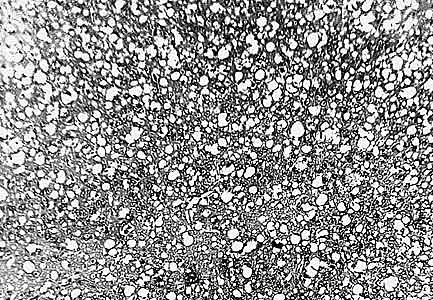
Микрофотография губкообразных изменений в коре большого мозга, вызванных прионами (болезнь Крейтцфельдта-Якоба).1- микровакуоли, 2 - погибающие нейроны с глиальными узелками, 3 - гипертрофия астроцитов, 4 - сморщенный нейрон, в котором уменьшились по объему цитоплазма и ядро, 5 - нейрон, в котором скопилось много липофусцина и произошло смещение ядра. Увел. ґ 400. (В.А.Зуев, И.А.Завалишин, В.М.Ройхель, 1999.)
Микрофотография конечной фазы губкообразных изменений в коре большого мозга: некоторые вакуоли слились и образовали более крупные полости, нейроны уже погибли. Увел. ґ 100.Довольно долго считалось, что все медленные болезни вызываются вирусами, однако постепенно, по мере накопления фактического материала, стала выделяться особая группа заболеваний человека и животных, возбудителей которых стали называть «необычными вирусами». Своеобразие этих болезней проявляется в избирательном поражении центральной нервной системы, что неуклонно приводит к губкообразному состоянию серого и/или белого вещества головного и спинного мозга при отсутствии воспалительной реакции. Выяснение патоморфологических и клинических особенностей этих необычных заболеваний позволило выделить их в отдельную группу медленных болезней под общим названием «трансмиссивные губкообразные энцефалопатии» (ТГЭ). По современной классификации, к ним относятся четыре болезни человека и шесть - животных (табл.1).
Все эти заболевания объединяет наличие единого (или близкого по свойствам) возбудителя, общность патогенеза, экспериментально показанная возможность переноса инфекции и воспроизведения клинической и патогистологической картины заболевания [6]. Эти выводы - итог многолетних исследований разных ученых - сформулировал американский биохимик С.Прузинер, получивший в 1997 г. Нобелевскую премию «за открытие прионов - нового биологического принципа инфекций». До этого все попытки обнаружить возбудителей ТГЭ заканчивались неудачей, хотя многое об их свойствах было известно. Прузинер назвал инфекционный агент прионом (частичная анаграмма от англ. proteinaceous infectious particles - белковоподобная инфекционная частица), а белок - РrP (от англ. prion protein).
Прионы, действительно, необычные патогены, они не способны вызвать острую форму инфекции. Связано это, видимо, с медленным процессом «перерождения» в зараженном организме неинфекционного клеточного белка РrРС (С от англ. cеll - клетка) - нормального компонента тканей млекопитающих, в том числе и человека, - в инфекционный прионный белок РrРSc (Sc от англ. scrapie - названия наиболее распространенной в природе прионной инфекции овец и коз).
Поскольку уже ни у кого не вызывает сомнений, что возбудителями ТГЭ могут быть только прионы, такие заболевания принято называть прионными [7]. Практический интерес к этим смертельно опасным инфекциям безусловно связан с событиями последнего времени и увеличивающейся вероятностью встречи с этими болезнями. Теоретический же интерес к проблеме обусловлен результатами молекулярно-биологических исследований прионов - новых и необычных возбудителей тяжелых заболеваний человека и животных, которые, как выяснилось, могут возникать не только в результате инфекции, но и спорадически и даже передаваться по наследству.
Структура прионных белков
Над выяснением структуры и химической природы возбудителей ТГЭ трудилось немало специалистов в течение, по крайней мере, полувека. В результате появилось большое количество разнообразных гипотез, многие из которых теперь имеют лишь историческое значение. Удача улыбнулась группе исследователей из Калифорнийского университета (США), работающих под руководством Прузинера. Главная их заслуга в том, что им удалось выяснить белковую природу прионов. Разработанная американскими учеными многоступенчатая система выделения исходного инфекционного материала позволила получить препараты, очищенные в 100-1000 раз. Агент оставался устойчивым к воздействию реагентов, инактивирующих нуклеиновые кислоты, что указывало на их отсутствие в его составе. Изучение очищенного препарата показало, что он обладает молекулярной массой около или меньше 50 кДа. В результате дальнейшей очистки приона выяснилось, что его основной компонент - мажорный белок с молекулярной массой 27-30 кДа, обозначаемый как РrР 27-30.
По физико-химической характеристике РrР 27-30 - сиалогликопротеин (олигосахаридсодержащий мембранный белок с остатками сиаловой кислоты, которые придают молекуле отрицательный заряд) и первый идентифицированный структурный компонент приона. Обнаружение РrР 27-30 на этапе развития инфекции, т.е. до появления патологических изменений в тканях, - свидетельство того, что этот белок не может быть вторичным продуктом патологической реакции. Так стало очевидным, что РrР 27-30 играет центральную роль в патогенезе заболевания.
При дальнейшем изучении прионов, выделенных из головного мозга зараженных скрепи животных, были обнаружены частицы в виде стержней диаметром 10-20 нм и длиной 100-200 нм. По ультраструктуре они напоминали амилоид (аномальный белок, который обычно образуется при хронических заболеваниях, например туберкулезе легких, костей и т.д.) и, видимо, представляли собой полимерную форму приона: каждый стержень содержал около тысячи молекул приона.
Важным шагом, имеющим как теоретическое, так и методическое значение, было получение антител при использовании в качестве антигена высокоочищенных прионов скрепи. В сыворотках кроликов, которым вводили РrР 27-30, обнаружены антитела не только к нему, но и к другим белкам, отличающимся более низкой молекулярной массой. Очевидно, эти белки либо обладают одинаковой антигенной детерминантой (областью антигена, комплементарной антителу) с РrР 27-30, либо они - продукт его расщепления. При помощи изготовленной антисыворотки с пероксидазной меткой удалось выявить локализацию прионов в определенных отделах головного мозга зараженных животных (табл.2). Согласно ранее полученным данным, структуры, связанные с меченой антисывороткой, обладали характеристикой амилоидных бляшек. Использование антисыворотки к синтетическому пептиду, соответствующему N-концевой части приона, позволило провести индикацию белка скрепи-ассоциированных фибрилл в головном мозге, селезенке и лимфатических узлах зараженных животных. При этом положительные результаты были получены на ранних этапах инкубационного периода.
Определение аминокислотной последовательности РrР 27-30 позволило в 1985 г. идентифицировать кодирующий его ген Prnp. Оказалось, что этот ген содержится в геномах не только инфицированных скрепи животных, но и здоровых. Соответственно мРНК для РrРС была выявлена в головном мозге и в других тканях как инфицированных, так и контрольных животных. Используя соответствующую антисыворотку, удалось показать, что в тканях незараженных животных содержится белок, родственный РrР 27-30, но отличающийся от него чувствительностью к обработке протеазой К.
Были изучены также некоторые другие характеристики прионов скрепи и болезни Крейтцфельдта-Якоба. В частности, было подтверждено предположение о том, что инфекционная частица агента содержит две молекулы РrР и что так называемые семейные формы (т.е. с наследственной предрасположенностью) прионных заболеваний связаны с конкретными мутациями в гене Prnp. Например, мутация, вызывающая замену пролина на лейцин в 102-м положении РrР, оказалась связана с развитием синдрома Герстманна-Штреусслера-Шейнкера, а замена аспарагиновой кислоты на аспарагин (мутация в 178-м кодоне) может быть связана как с болезнью Крейтцфельдта-Якоба, так и со смертельной семейной бессонницей. К сегодняшнему дню известно уже о 20 мутациях в гене Prnp человека, связанных с семейными формами прионных заболеваний.
Физико-химические свойства
Во второй половине 90-х годов, когда уже была определена аминокислотная последовательность РrР и выявлен ген Prnp, начались интенсивные поиски причин патогенности прионов. С помощью современных методов молекулярно-генетического анализа были получены новые данные о возможных вариантах состава и конформации (укладки) полипептидной цепи РrР. В частности, было установлено, что конверсия нормального прионного белка в его инфекционную изоформу - посттрансляционный процесс [8]. Анализ вторичной структуры РrРSc показал, что этот переход характеризуется большими структурными изменениями самого приона. Клеточный белок содержит 42% a-спиралей и почти не содержит b-тяжей (всего около 3%), в то время как в его инфекционной форме выявляется 30% a-спиралей и 43% b-тяжей [9]. В экспериментальных исследованиях было подтверждено, что обработка неинфекционного белка реагентами, снижающими образование b-тяжей, также приводила к уменьшению инфекционности перерожденного приона. Одновременно снижалась и устойчивость РrРSc к действию протеазы К, чувствительность к которой считается маркером, отличающим РrРC от РrРSc.
Превращение нормального белка в патогенный, судя по всему, происходит путем белок-белковых взаимодействий, при этом не имеет значения, попадает РrРSс в организм извне или возникает в нем спонтанно (в случае спонтанных и наследственных прионных болезней). Как это происходит, уверенно сказать пока нельзя, однако в настоящее время предлагаются две модели, описывающие это превращение: «гетеродимерная» и «полимеризационная». Согласно первой, прионное состояние присуще мономеру белка РrР, который катализирует конформационный переход молекулы РrРC в форму РrРSс [10]. После того как нормальный белок приобретает прионные свойства, димер диссоциирует, и две освободившиеся молекулы РrРSс могут вновь взаимодействовать с очередными молекулами РrРC. Процесс напоминает цепную реакцию и может протекать довольно быстро, однако остается необъясненным механизм образования амилоидных бляшек. Вторая модель с этой точки зрения более перспективна, поскольку рассматривает прион как упорядоченный полимер РrР. Процесс же его конформационой перестройки, согласно этой теории, напоминает кристаллизацию, которую запускает олигомер РrРSс [11]. Отложения белка РrРSс (бляшки) в тканях мозга заболевших людей обычно содержат нитевидные агрегаты этого белка, что свидетельствует об упорядоченной его полимеризации.
Таким образом, в результате разносторонних исследований были получены и систематизированы имеющие принципиальное значение данные о структуре и физико-химических свойствах прионных белков. Анализ этих сведений создал необходимые предпосылки для дальнейшего углубленного изучения биологических особенностей прионных белков и механизма развития вызываемых ими заболеваний людей и животных.
Биологические особенности
Несмотря на обилие результатов в изучении прионных белков, роль PrPC в живом организме до конца не известна. Ясно одно – этот белок жизненно необходим и эволюционно консервативен, поскольку обнаружен у многих млекопитающих, птиц и даже у низших эвкариот. Так, при анализе первичной структуры РrРС было выявлено, что 80% последовательностей РrРС у разных видов животных идентичны, исключение составлял лишь куриный РrРС, где идентичность последовательностей по отношению к другим видам составляла всего 30%. Тем не менее 24 аминокислотные последовательности, располагающиеся между 112-м и 135-м аминокислотными остатками, содержатся в геномах всех млекопитающих, а также кур.
В экспериментах на трансгенных мышах, гомозиготных по потере гена Prnp, было показано, что эти животные после рождения росли «нормальными», но спустя 70 недель у них развились прогрессирующие симптомы атаксии, в частности нарушилась моторная координация вследствие экстенсивной потери клеток Пуркинье (крупных нейронов коры мозжечка). Помимо этого, установлено, что PrPC играет важную роль в регуляции циркадианных (суточных) ритмов, возможно, участвует в активации лимфоцитов, а также выполняет функции трофического фактора для некоторых популяций нейронов. Сохранность PrPC имеет значение для реализации нормальной функции синапсов.
В последние годы появились данные, свидетельствующие о роли клеточного белка в регуляции сна, и более того - возникновение смертельной семейной бессонницы связывают с нарушением нормальной функции этого белка [12]. В исследованиях in vitro было показано, что PrPC вовлекается в процессы регуляции содержания внутриклеточного Са2+ в нейронах [13]. Уже доказанным можно считать и значение нормального клеточного приона в сохранении резистентности нейронов и астроцитов к окислительному стрессу, и участие этого белка в метаболизме меди в головном мозге [14]. А совсем недавно были получены данные об участии PrPC в трансдукции сигналов в нервной ткани [15]. Этот список можно было бы продолжить, но и так ясно, что представления о биологической значимости PrPC в последние годы существенно расширились.
Теперь стало известно, что PrPC синтезируется в эндоплазматической сети и довольно быстро деградирует (всего за 5-6 часов). Синтезированный PrPC, проходя через аппарат Гольджи, транспортируется на поверхность клетки, где он связывается с гликофосфатидилинозитолом и в дальнейшем переносится вдоль аксона при помощи быстрого и активного (антероградного) транспорта. В отличие от PrPC инфекционный прионный белок первично аккумулируется в клетках, накапливаясь в цитоплазматических везикулах. Дальнейшее его накопление в синаптических структурах и связанная с этим дезорганизация синапсов, очевидно, становится причиной глубоких неврологических дефектов и деменции.
В изучении патогенеза и эпидемиологии прионных болезней существует много белых пятен. К ним относятся, в частности, пути передачи заболевания в природе, взаимосвязь болезней человека и животных, определение «входных ворот» инфекции [16]. Судя по всему, наиболее вероятный путь передачи заболевания - алиментарный. Правомерность этого вывода была косвенно подтверждена, когда для искоренения куру на о.Новая Гвинея достаточно было запретить обычай ритуального каннибализма. Доказано это было и в экспериментах, в которых животным (в том числе и приматам) скармливали высоко инфицированные субстраты - ткани головного мозга больных животных.
Получение новых данных позволило заключить, что прионные болезни - нейродегенеративные, в возникновении которых фундаментальную роль играют конформационные изменения прионов, а сам механизм развития болезни беспрецедентен.
* * *
Результаты исследований, проведенных в последние 10-15 лет, позволили с новых позиций подойти к вопросу о природе агентов ТГЭ, а сумма полученных новых знаний о прионах послужила основанием для оптимистического высказывания Прузинера: «Эра черного ящика биологии скрепи и болезни Крейтцфельдта-Якоба, возможно, подходит к концу». Хочется надеяться, что проблема медленных прионных болезней действительно будет решена в скором будущем. Успехи в этой области очевидны, о чем свидетельствует хотя бы то, что на протяжении 20 лет Нобелевский комитет дважды отмечал ученых за достижения в одной и той же области медико-биологических исследований.
Литература
1. Кемпбелл П.Н. // Вопр. биол., мед. и фармац. химии. 1998. №4. С.34-40.
2. Hill A., Debruslais M., Joiner M. et al. // Nature. 1997. V.389. P.448-450.
3. Calza L., Manfredi R., Chiodo F. // Recenti Prog. Med. 2001. V.92. P.140-149.
4. Sigurdsson P. // British Veterinary J. 1954. V.110. P.341-354.
5. Зуев В.А. Медленные вирусные инфекции человека и животных. М., 1988.
6. Prion Biology and Diseases / Ed. S.B.Prusiner. N.Y., 1999.
7. Зуев В.А., Завалишин И.А., Ройхель В.М. Прионные болезни человека и животных. М., 1999.
8. Pan K., Baldwin M., Nguyen J. et al. // Proc. Natl. Acad. Sci. 1993. V.90. P.10926-10966.
9. Smith C., Collinge J. // Essay Biochem. 1995. V.29. P.157-174.
10. Cohen F.E., Pan K.M., Huang Z. et al. // Science. 1994. V.264. P.530-531.
11. Jarrett J.T., Lansbury P.T. // Cell. 1993. V.73. P.1055-1058.
12. Tobler I., Deboer Т., Fisher M. // J. Neurosci. 1997. V.17. P.1869-1879.
13. Herms J., Tings Т., Dunker S. et al. // J. Neurobiol. Dis. 2001. V.8. P.324-330.
14. Brown D. // J. Brain Res. Bull. 2001. V.55. P.165-173.
15. Martins V., Mercadante A., Cabral A. et al. // Braz. J. Med. Biol. Res. 2001. V.34. P.585-595.
16. Ройхель В.М. Патогенез и диагностика некоторых медленных прионовых нейроинфекций: Автореферат на соискание ученой степени доктора медицинских наук. М., 1997.
17. Brown P. // J. Microsci Res. Tech. 2001. V.54. P.71-80.