№ 8, 2003 г.
© Л.Л.КиселевКАК ОСТАНОВИТЬ СИНТЕЗ БЕЛКОВ?
Л.Л. Киселев
Лев Львович Киселев, академик РАН и Европейской академии (Academia Europaea),
заведующий лабораторией Института молекулярной биологии им.В.А.Энгельгардта РАН.Благодаря достижениям молекулярной биологии последних двух десятилетий мы знаем, как инициируется белковый синтез на рибосомах (клеточных частицах, в которых протекает трансляция, т.е. синтез белков) и как происходит удлинение полипептидной цепи (элонгация). Однако завершение синтеза (терминация) оставалось вне поля зрения исследователей, хотя еще с конца 1960-х годов известны два основополагающих факта. Во-первых, синтез прекращается, когда в участке А малой субъединицы рибосомы появляется стоп- (нонсенс-)кодон. Это один из трех тринуклеотидов (УАА, УАГ, УГА), которые не кодируют аминокислот, а служат сигналом для окончания элонгации (рис.1). Во-вторых, “считывание” стоп-кодонов в рибосоме нуждается в особых белках, названных факторами освобождения полипептидных цепей (release factors, сокращенно RF). По свойствам и роли в клетке эти белки подразделяются на два класса. У бактерий известны два фактора терминации класса-1 - RF1 (узнающий стоп-кодоны УАА и УАГ) и RF2 (узнающий УАА и УГА), у архебактерий - aRF1, у эвкариот - eRF1 (узнающие все три стоп-кодона).
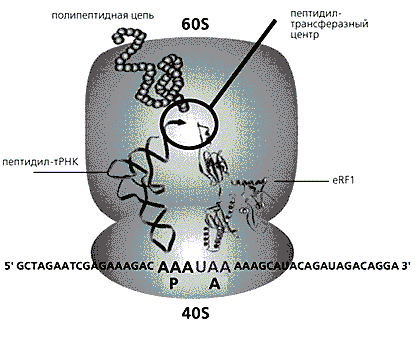
Рис. 1. Схема терминации белкового синтеза у эвкариот.По мере того, как молекула мРНК продвигается сквозь рибосому (состоящую из 60S и 40S субчастиц), ее нуклеотидная последовательность переводится в аминокислотную. Присутствие стоп-кодона UAA и eRF1 в А-участке рибосомы приводит к остановке синтеза - расщеплению пептидил-тРНК (освобождению полипептидной цепи).К концу 80-х годов аминокислотная последовательность многих бактериальных факторов стала известна, а вот с eRF1 произошла драматическая история, о которой я уже рассказывал [1]. Американские исследователи в результате методических ошибок приписали ему структуру совсем другого белка. Обнаружив это [2], мы смогли расшифровать истинную последовательность аминокислот eRF1 [3], что позволило идентифицировать похожие на него архебактериальные факторы aRF1. При этом выяснилось, что eRF1 резко отличается по структуре от ранее известных RF1 и RF2, и мы предположили, что факторы терминации бактерий и эвкариот произошли от разных предковых молекул [3]. Такая гипотеза вызвала резкую критику со стороны некоторых исследователей. Однако дальнейшее изучение большого числа структур [4], а также рентгеноструктурный анализ кристаллов eRF1 (выполненный в Англии) и RF2 (в Дании) полностью подтвердили нашу правоту.Структуру факторов класса-2 бактериального RF3 [5] и эвкариотического eRF3 [6] установили почти одновременно. Они оказались ферментами, которые катализируют внутри рибосом гидролиз гуанозинтрифосфата (ГТФ), не участвуют в терминации (расщеплении конечного продукта белкового синтеза, пептидил-тРНК), но способствуют освобождению из рибосом факторов класса-1 [7, 8].
Ключевая роль трипептида GGQ
Бактериальные и эвкариотические факторы класса-1, несмотря на несходство своих первичных структур, имеют один общий элемент - трипептид глицил-глицил-глютамин (сокращенно GGQ). Он присутствует во всех белках класса-1, а их сейчас известно для бактерий, архебактерий и эвкариот уже около 100 (рис.2). Обнаружив совместно с коллегами из центра по вирусологии и биотехнологии “Вектор” этот универсальный мотив, мы предположили, что он ответствен за химическую стадию терминации (расщепление пептидил-тРНК) [9]. Это означало, что его изменение приведет к потере активности фактора, но не помешает ему связываться с рибосомой, поврежденный белок превратится в ингибитор терминации. Сам трипептид GGQ должен находиться в большой субчастице рибосомы, в области ее пептидилтрансферазного центра. При экспериментальной проверке все эти следствия полностью подтвердились.
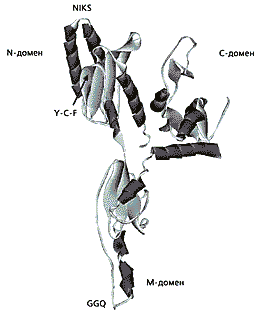
Рис. 2. Схема трехмерной структуры eRF1 человека
по данным рентгеноструктурного анализа [12].N-домен узнает стоп-кодон, M-домен находится в пептидилтрансферазном центре рибосом и участвует в гидролизе пептидил-тРНК, С-домен связывается с фактором класса-2 eRF3. Трипептид GGQ находится на одном из концов молекулы eRF1, образуя гибкую петлю. Такое пространственное расположение обеспечивает контакт eRF1 с пептидил-тРНК, которая располагается в соседнем с молекулой участке (Р).
Таким образом, удалось показать, что GGQ служит активным центром факторов класса-1 (у прокариот и эвкариот) и, располагаясь в пептидилтрансферазном центре, катализирует расщепление эфирной связи в пептидил-тРНК. Интересно, что ключевая роль этого трипептида в терминации обеспечивается в первую очередь двумя глициновыми остатками, а не глютамином [10-15]. Возможно, что отсутствие у глицина боковой группы освобождает пространство для молекулы воды, участвующей в гидролизе пептидил-тРНК, поскольку замены глициновых остатков на более объемные неизменно ведут к утрате активности у мутантных RF.
Как факторы узнают стоп-кодоны?
Фактор класса-1 катализирует гидролиз, только если в А-участке малой субъединицы присутствует один из трех стоп-кодонов. Иными словами, внутри рибосомы сигнал от стоп-кодона (находящегося в малой субчастице) к пептидилтрансферазному центру (расположенному в большой рибосомной субчастице) передается через GGQ-мотив. Как такая передача может осуществляться? Согласно одной гипотезе, стоп-кодон специфически связывается с участком малой рибосомной РНК, а фактор играет лишь вспомогательную роль. Согласно другой, стоп-кодон взаимодействует с “узнающим” трипептидом фактора класса-1. В первом случае главную роль в передаче сигнала играет рибосома, а во втором - фактор класса-1. На основании совместных опытов с французскими [16] и японскими [17] исследователями мы отдали предпочтение второй гипотезе. Оказалось: если в пробирке создать “гибрид” из рибосомы, умеющей “считывать” все три стоп-кодона, и фактора класса-1, выделенного из организма, где используются только один или два стоп-кодона, то результат будет зависеть от природы фактора. Отсюда можно предполагать прямой контакт eRF1 со стоп-кодоном в рибосоме. Действительно, используя синтезированные триплеты с химически модифицированными группами, мы совместно с парижскими и новосибирскими коллегами доказали, что они образуют ковалентную связь с молекулой eRF1 внутри рибосомы [18, 19]. Это возможно лишь в том случае, если они сближены внутри рибосомы. Аналогичные результаты были получены другими исследователями для факторов класса-1 и рибосом бактерий. Следовательно, факторы класса-1 должны иметь участок, специфически узнающий в рибосоме стоп-кодоны мРНК.
Недавно такой участок удалось найти у RF1 и RF2 бактерий [20]. Это были трипептиды, но разные (поскольку RF1 узнает УАГ, а RF2 - УГА, а УАА узнают оба фактора). Пока не показано, находятся ли они в непосредственном контакте со стоп-кодоном в рибосоме. Однако результат ничего не дал для понимания терминации у эвкариот, поскольку первичные и пространственные структуры этих факторов класса-1 у двух царств живой природы устроены по-разному.
Исходя из того, что расстояние между стоп-кодоном и пептидилтрансферазным центром рибосом составляет примерно 75 Е (это вытекает из формы и размеров тРНК и данных рентгеноструктурного анализа рибосом прокариот), узнающий участок может располагаться либо на N-концевом, либо на С-концевом доменах. Второе предположение отпадает сразу же, поскольку удаление С-домена не мешает узнаванию: такой укороченный белок в пробирке в полной мере сохраняет свою активность [21]. Значит, стоп-кодон должен узнаваться N-доменом, что подтверждается опытами с мутантным [22, 23] и “гибридным” eRF1 [17], а также данными других исследователей.
Однако пока не ясно, где именно находится этот участок, так как размер N-домена достаточно велик. В поисках ответа мы проанализировали аминокислотную последовательность факторов eRF1 из самых разных организмов (по данным из общедоступных банков). Два пептидных фрагмента, Асн-Иле----Лиз-Сер (NIKS) и Тир-Цис---Фен (Y-C-F) *, расположены в петлях N-домена (полностью или частично), что облегчает им контакт со стоп-кодоном в рибосоме. Присутствие в пептидах консервативных и полуконсервативных аминокислотных остатков также не противоречит гипотезе о их возможной роли как узнающих стоп-кодон. Однако нужны были прямые экспериментальные доказательства, которые мы получили с помощью направленного мутагенеза.
* Серия тире означает, что между этими инвариантными аминокислотами находятся другие, варьирующие.Мы заменили в тетрапептиде NIKS каждую аминокислоту и посмотрели, как это сказывается на функциональной активности eRF1 человека. Опыты, поставленные в пробирке, дали вполне четкие результаты [22]. Замена изолейцина (I) весьма серьезно уменьшает активность мутантного белка; эффект от замены лизина (К) гораздо мягче; крайние аминокислоты аспарагин (N) и серин (S) занимают промежуточные положения. В целом, фрагмент NIKS безусловно функционально важен, но утверждать на основании наших данных, что именно он узнает весь стоп-кодон, вряд ли правильно.Совершенно иную картину мы получили, когда анализировали участок Y-C-F [23]. При замене цистеина (С) или фенилаланина (F) на другие аминокислоты ответ на стоп-кодон УГА почти не менялся, а на УАА и УАГ падал очень сильно. Более того, если к каждой из этих мутаций добавить еще по одной - по аспарагину (N), стоящему между C и F, то eRF1 человека становится похожим на eRF1 реснитчатых простейших. У них фактор узнает только УГА, а два других триплета (УАА и УАГ) превращаются в значащие, кодирующие аминокислоты. Такие белки, узнающие только один стоп-кодон, называют унипотентными, в отличие от омнипотентных eRF1 высших эвкариот.
Итак, направленное изменение структуры eRF1 в области Y-C-F-петли привело к потере узнающей способности фактора в отношении двух стоп-кодонов, сохранив ее в отношении третьего, УГА. Значит, и цистеиновый, и фенилаланиновый остатки, безусловно, участвуют в узнавании стоп-кодонов, хотя пока не известно, прямо или косвенно. Поскольку у двух триплетов, УАА и УАГ, в отличие от УГА, общий элемент - второй нуклеотид (аденин), вполне возможно, что именно в его узнавании участвуют цистеин и фенилаланин.
Однако такой вывод противоречит тому известному обстоятельству, что в организмах с вариантными генетическими кодами (реснитчатых простейших) узнается только часть стоп-кодонов, а эти аминокислоты тем не менее присутствуют в структуре их факторов. Чтобы объяснить такую ситуацию, мы предложили концепцию негативных элементов, которые препятствуют у реснитчатых участию цистеина и/или фенилаланина в узнавании стоп-кодонов (возможно, аденина во втором положении). Действительно, если посмотреть на аминокислоты, окружающие цистеин и фенилаланин слева и справа, видно (рис.3), что они у организмов с универсальным генетическим кодом (высших эвкариот) отличаются от таковых с вариантными кодами. Некоторые из этих аминокислот могли бы выполнять роль таких негативных элементов. Разумеется, наше предположение требует экспериментальной проверки. Однако нечто подобное достаточно хорошо изучено на примере тРНК и аминоацил-тРНК-синтетаз, где в тРНК отдельные нуклеотиды как раз и служат негативными элементами.
Рис. 3. Сравнение аминокислотных последовательностей (в однобуквенном аминокислотном коде) факторов класса-1 в местах расположения инвариантного трипептида GGQ (из базы данных NCBI-Eutrer Proteins). Очевидно, что этот элемент присутствует во всех белках класса-1 от эвкариот до архебактерий.Превращение омнипотентного фактора (eRF1 человека) в унипотентный (характерный для некоторых простейших эвкариот, узнающий только УГА), осуществленное с помощью точечного мутагенеза, скорее всего имитирует процесс, происходивший в живой природе десятки миллионов лет назад. Согласно воззрениям эволюционистов, организмы с вариантными кодами произошли от организмов с универсальным (стандартным) генетическим кодом, а не наоборот. Иными словами, раньше факторы терминации реснитчатых умели узнавать все стоп-кодоны, а потом “разучились”. Можно ли каким-либо способом убедиться, что такой потенциал у них сохранился? Оказалось, что универсальность узнавания можно вернуть, причем крайне просто - изменив температуру культивирования организма [17]. Этот опыт вместе с другими подтверждает два предположения: во-первых, между факторами терминации из организмов с универсальным и вариантными кодами нет пропасти (при некоторых обстоятельствах они взаимно превращаемы). Во-вторых, если нейтрализовать действие негативных элементов (в данном случае изменением температуры), узнаются все стоп-кодоны.Согласно гипотезе белкового антикодона, предложенной для прокариот, трипептиды PAT (Про-Ала-Тре) в RF1 или SPF (Сер-Про-Фен) в RF2 узнают по два стоп-кодона из трех [20]. Применима ли концепция белкового антикодона (т.е. короткой линейной последовательности аминокислот, узнающей тринуклеотид) к eRF1?
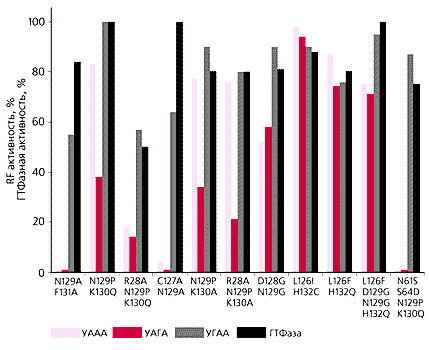
Наши опыты скорее говорят об обратном [23]. Превращение омнипотентного фактора в унипотентный удалось достичь, не только меняя цистеин или фенилаланин, но и другие аминокислоты в двух минидоменах NIKS и Y-C-F одновременно. Изменения по отдельности в каждом из них недостаточны для подобного эффекта (рис.4). Значит, два минидомена, разнесенных в пространстве, при узнавании стоп-кодонов взаимодействуют друг с другом. Это уже не линейная непрерывная последовательность аминокислот, как предполагается для RF1 и RF2. Видимо, помимо глубоких отличий в структуре факторы класса-1 прокариот и эвкариот различаются и по механизму узнавания.
Рис. 4. Функциональная активность мутантов eRF1 человека в сравнении с исходным фактором по двум тестам: освобождению полипептида и тройной ГТФазной активности (в тесте использовали тетрапептиды для улучшения их связывания с рибосомой). Мутации указаны под столбиками в однобуквенном аминокислотном коде; слева - исходная аминокислота (числа обозначают ее порядковый номер), справа - мутантная. Видно, что с помощью двойных и четверных мутаций омнипотентный фактор превращается в унипотентный (узнает только УГА).Новый методИзучение молекулярного механизма терминации нуждается в адекватных экспериментальных подходах. Методы, которыми пользуются многие исследователи, для этих целей уже недостаточно совершенны. Сегодня необходим метод, который объединял бы условия опыта in vivo, существующие в клетке, и in vitro - в пробирке, когда экспериментатор может менять условия опыта по своему желанию. Решая такую непростую задачу [24], мы опирались на показанную нами вместе с французскими исследователями in vitro конкуренцию за стоп-кодоны между eRF1 и супрессорными тРНК [25], которые могут “читать” стоп-кодоны как значащие (кодирующие ту или иную аминокислоту). Этот принцип позволяет оценивать количественно свойства как eRF1 (и, разумеется, его мутантов), так и супрессорных тРНК. В качестве матрицы, которая связывалась с рибосомами, мы использовали копию гена люциферазы (фермента, воздействие которого на субстрат приводит к испусканию кванта света). Если матрица “считывается” полностью, образуется активный белок, который заставляет люциферин (субстрат) хорошо светиться в рибосомной системе, снабженной всеми необходимыми компонентами. Если же в матрицу вставить стоп-кодон, синтез полипептида обрывается, и свечения субстрата нет. Добавляя в такую систему eRF1, супрессорные тРНК и их смеси, можно “по заказу” менять интенсивность синтеза люциферазы и по свечению измерять ее активность. Эта же система годится и для изучения свойств и активности eRF3.
Теперь мы имеем такой метод оценки свойств eRF1, который в дальнейшем позволит по-новому мерить кинетику терминации и осуществлять варианты анализа, нереальные при использовании традиционных подходов.
Возможности молекулярной терапии
Феномен конкуренции eRF1 и супрессорных тРНК [24, 25] натолкнул нас еще на одну идею, относящуюся к биомедицине или, как сейчас модно говорить, к молекулярной медицине. Многие наследственные болезни связаны с тем, что в результате повреждения гена один из значащих триплетов превращается в незначащий (такие мутации называют нонсенс-мутациями). Этот стоп-кодон, попадая в мРНК, приводит к синтезу укороченного белка, который, как правило, биологически неактивен.
Теоретически существуют два принципиально разных пути устранения такого дефекта. Первый - генотерапия, т.е. замещение поврежденного гена нормальным, не содержащим в кодирующей области стоп-кодона. Методы генотерапии сейчас активно разрабатываются, но, к сожалению, до сих пор без особого успеха. Второй путь связан с исправлением генного продукта - белка.
Как можно вылечить больной белок? Очевидно, нужно не дать сработать стоп-кодону внутри мРНК (т.е. обеспечить достройку полипептидной цепи до конца), что возможно двумя способами. Первый - блокировать каким-либо образом eRF1 (например, добавив к нему антитела или соответствующий ингибитор). Терминация будет ослаблена, и в действие вступят супрессорные тРНК, которые в небольших количествах всегда присутствуют в клетках высших организмов. Второй - внести в систему супрессорные тРНК, которые за счет конкуренции вытеснят eRF1 из А-участка рибосомы, а стоп-кодон будет прочитан как значащий. Это, скорее всего, приведет к синтезу полноразмерного белка с нормальной активностью.
Оба подхода реализованы нами экспериментально. С помощью популярного метода Selex мы совместно с учеными США получили набор коротких молекул РНК (аптамеров), которые блокируют активность фактора eRF1 in vitro [26]. Есть надежда, что если такие ингибиторы-аптамеры ввести в клетки больного, терминация будет ослаблена и больной белок вылечится.
Совместно с Институтом акушерства и гинекологии им.Д.О.Отта РАМН в Санкт-Петербурге [27] на мышах, у которых поврежден ген, кодирующий белок дистрофин, мы показали, что после введения гена супрессорной тРНК в мышечные клетки в них появляется дистрофин. Иными словами, когда в клетках возрастает концентрация супрессорной тРНК, eRF1 вытесняется из А-участка рибосомы и часть полипептидных цепей считывается до конца, без обрыва на месте стоп-кодона.
Таким образом, способы воздействия на терминацию в принципе могут стать основой молекулярной терапии всех наследственных и ненаследственных дефектов белков, связанных с преждевременным завершением синтеза. Конечно, предстоит преодолеть еще много трудностей на этом пути, и до лечения больных пока далеко. Однако впервые в истории медицины появились, по крайней мере идеи о том, как можно подступиться к болезням, которые до сих пор считались неизлечимыми.
Помимо своей основной функции - участия в передаче сигнала от стоп-кодона к пептидилтрансферазному центру рибосомы eRF1 играют еще одну важную роль: они активируют ГТФазную активность фактора eRF3 класса-2. Факторы eRF1 и eRF3 человека взаимодействуют друг с другом своими С-концевыми участками, образуя достаточно прочные комплексы, они обнаружены как in vitro, так и in vivo, причем независимо от присутствия рибосом и стоп-кодонов [21, 28, 29]. У эвкариот eRF3 катализирует расщепление ГТФ только при одновременном присутствии рибосом и eRF1 [7]. Такая же ГТФазная активность отмечена и для бактериальных факторов RF3. Однако если eRF1 и eRF3 in vitro дают достаточно прочный комплекс, то у прокариот такого комплекса пока никто не получил.
После обнаружения этого феномена появились две гипотезы [30] о возможной роли факторов класса-1 в отношении факторов класса-2. Согласно первой, она заключается в активации фермента путем “достройки” его активного центра. Вторая гипотеза постулирует, что факторы класса-1, связываясь с ГТФазами, резко ускоряют обмен ГТФ-ГДФ на ферменте, что увеличивает каталитическую активность. Первая гипотеза еще нуждается в экспериментальной проверке [31], вторая - экспериментально подтверждена для бактериального eRF3 [32].
Подведем краткие итоги 10-летнего изучения терминации белкового синтеза у эвкариот. * * *
Раскрыта структура (первичная, вторичная, пространственная) белка eRF1 - фактора терминации класса-1.
Установлено, что он определяет специфичность считывания стоп-кодонов, взаимодействуя своим N-концевым доменом со стоп-кодоном в рибосоме.
Выявлены аминокислоты, существенные для функций eRF1 в рибосоме.
Доказано, что универсальный инвариантный трипептид Гли--Гли-Глн играет решающую роль при расщеплении пептидил-тРНК. Феномен конкуренции между eRF1 и супрессорными тРНК за стоп-кодон в А-участке рибосомы лег в основу нового метода определения активности фактора и нового подхода к лечению наследственных болезней человека, связанных с нонсенс-мутациями.
Получены избирательные ингибиторы eRF1 (РНК-аптамеры).
Установлены первичная структура и биологическая функция фактора терминации класса-2 - eRF3.
Обнаружен контакт между факторами двух классов eRF1 и eRF3 in vitro и in vivo и раскрыт механизм их взаимодействия через С-концевые домены этих белков.
Сформулирована гипотеза о биологической роли eRF3 в клетках.
На молекулярном уровне определены различия между терминацией белкового синтеза у прокариот и эвкариот.
Достижения российской физико-химической биологии в этой области полностью признаны мировым научным сообществом, что легко проследить по недавним обзорам этой проблемы [30, 31, 33-36].
Литература
1. Киселев Л.Л. Детективная история из жизни молекулярных биологов // Природа. 1993. ?7. С.91-95.
2. Frolova L., Dalphin M., Justesen J. et al. // EMBO J. 1993. V.12. P.4013-4019.
3. Frolova L., Goff X. le., Rasmussen H.H. et al. // Nature. 1994. V.372. P.701-703.
4. Киселев Л.Л., Опарина Н.Ю., Фролова Л.Ю. // Молекуляр. биология. 2000. Т.34. С.427-441.
5. Grentzmann G., Brechemier-Baey D., Heurgue V. et al. // Proc. Natl. Acad. Sci. USA. 1994. V.91. P.5848-5852.
6. Zhouravleva G., Frolova L., Goff X.le et al. // EMBO J. 1995. V.14. P.4065-4072.
7. Frolova L., Goff X.le, Zhouravleva G. et al. // RNA. 1996. V.2. P.334-341.
8. Freistroffer D.V., Pavlov M.Y., MacDougаll J. et al. // EMBO J. 1997. V.16. P.4126-4133.
9. Frolova L.Y., Tsivkovskii R.Y., Sivolobova G.F. et al. // RNA. 1999. V.5. P.1014-1020.
10. Сеит-Неби А., Фролова Л., Иванова Н. и др. // Молекуляр. биология. 2000. Т.34. С.899-900.
11. Seit-Nebi A., Frolova L., Justesen J., Kisselev L. // Nucl. Acids Res. 2001. V.29. P.3982-3987.
12. Song H., Mugnier P., Das A.K. et al. // Cell. 2000. V.100. P.311-321.
13. Zavialov A.V., Mora L., Buckingham R.H. et al. // Mol. Cell. 2002. V.10. ?4. P.789-798.
14. Mora L., Heurgue-Hamard V., Champ S. et al. // Mol. Microbiol. 2003. V.47(1). P.267-275.
15. Janzen D.M., Frolova L., Geballe A. // Mol. Cell. Biol. 2002. V.22. ?24. P.8562-8570.
16. Kervestin S., Frolova L., Kisselev L. et al. // EMBO Rep. 2001. V.2. P.680-684.
17. Ito K., Frolova L., Seit-Nebi A. et al. // Proc. Natl. Acad. Sci. USA. 2002. V.99. P.8494-8499.
18. Chavatte L., Frolova L., Kisselev L. et al. // Eur. J. Biochem. 2001. V.268. P.2896-2904.
19. Bulygin K.N., Repkova M.N, Ven’yaminova A.G. et al. // FEBS Letters. 2002. V.514. P.96-101.
20. Ito K., Uno M., Nakamura Y. // Nature. 2000. V.403. P.680-684.
21. Frolova L.Y., Merkulova T.I., Kisselev L.L. // RNA. 2000. V.6. P.381-390.
22. Frolova L., Seit-Nebi A., Kisselev L. // RNA. 2002. V.8. P.129-136.
23. Seit-Nebi A., Frolova L., Kisselev L. // EMBO Rep. 2002. V.3. P.881-886.
24. Мазур А.М., Холод Н.С., Сеит-Неби А.С. и др. // Молекуляр. биология. 2002. Т.36. С.129-135.
25. Drugeon G., Jean-Jean O., Frolova L. et al. // Nucleic Acids Res. 1997. V.25. P.2254-2258.
26. Carnes J., Frolova L., Zinnen S. et al. // RNA. 2000. V.6. P.1468-1479.
27. Киселев А.В., Остапенко О.В., Рогожкина Е.В. и др. // Молекуляр. биология. 2002. Т.36. С.43-47.
28. Frolova L., Simonsen J., Merkulova T. et al. // Eur. J. Biochem. 1998. V.256. P.36-44.
29. Merkulova T.I., Frolova L.Y., Lazar M. et al. // FEBS Lett. 1999. V.443. P.41-47.
30. Buckingham R., Grentzmann G., Kisselev L. // Mol. Microbiol. 1997. V.24. P.449-456.
31. Kisselev L.L., Ehrenberg M., Frolova L.Yu. // EMBO J. 2003. V.22. ?2. P.175-182.
32. Zavialov A.V., Buckingham R.H., Ehrenberg M. // Cell. 2001. V.107. P.115-124.
33. Kisselev L.L., Buckingham R.H. // Trends Biochem. Sci. 2000. V.25. P.561-566.
34. Ehrenberg M., Tenson T. // Nature Struct. Biol. 2002. V.9. P.85-87.
35. Janzen D.M., Geballe A.P. Cold Spring Harbor Symp. Quant. Biol. Cold Spring Harbor, 2001. P.459-467.
36. Kisselev L.L. Structure. 2002.
Июль 2003